文献速递 | 中性粒细胞弹性蛋白酶(NE)促进胸主动脉夹层形成

胸主动脉夹层(TAD)是一种危及生命的急性心血管疾病,其发病机制涉及炎症反应与血管平滑肌细胞(SMC)表型转换等多个病理过程。近期研究表明,中性粒细胞弹性蛋白酶(NE)在TAD发生发展中可能起着关键作用。2023年8月,上海交通大学医学院附属新华医院张力教授团队在《Arteriosclerosis, Thrombosis, and Vascular Biology》(IF=7.4,Q1)发表的研究进一步揭示,NE及其下游靶点TBL1x通过调控炎症细胞迁移和SMC表型转换,推动TAD进展,为该疾病提供了新的潜在治疗靶点。支持这一观点的是,与正常主动脉相比,断裂的主动脉组织及急性TAD患者主动脉中NE蛋白与基因表达均显著上升,且患者血清NE水平也明显升高。临床数据分析显示,血清NE水平对急性TAD具有良好的诊断价值,其受试者工作特征曲线下面积达0.7415;当临界值取2.783纳克/毫升时,鉴别急性TAD患者与健康人的特异性为81%,灵敏度为72%。这些结果共同提示,NE不仅参与TAD的病理生理机制,也可能作为辅助诊断的生物标志物,为早期识别与干预高危TAD患者提供新方向。
文章亮点
NE缺失可降低胸主动脉夹层(TAD)发生率并提高动物存活率。
药物抑制NE可预防TAD的发生与发展。
NE通过蛋白水解切割TBL1x蛋白(一种含F-box样/WD重复序列的蛋白)来促进TAD发展。
NE通过调节TBL1x-MECP2信号轴,在TAD病理条件下调控平滑肌细胞的表型转换。
NE通过调节TBL1x-LTA4H信号轴,介导炎症细胞的跨内皮迁移。
rId5
目的:胸主动脉夹层(TAD)是一种致命的主动脉疾病,目前缺乏有效的药物治疗。越来越多的证据表明,中性粒细胞弹性蛋白酶(NE)在血管疾病中发挥作用。本研究旨在探讨NE在TAD中的致病作用及其分子机制。
方法:使用β-氨基丙腈单富马酸盐(BAPN)诱导小鼠TAD模型。通过NE基因敲除小鼠、NE药理抑制剂GW311616A以及腺相关病毒2(AAV2)介导的体内基因转移等技术,探讨NE及其靶基因在TAD形成中的作用。通过多种功能实验和生化分析,阐明NE在TAD中的细胞与分子机制。
结果:在BAPN诱导的TAD模型及急性TAD患者中,主动脉NE基因表达和血浆NE活性均显著升高。NE缺失可预防BAPN诱导的TAD发生与发展,而给予抑制剂GW311616A也能改善TAD的形成与进展。在NE缺失小鼠中,观察到中性粒细胞胞外诱捕网(NETs)、炎症细胞以及基质金属蛋白酶(MMP)-2/9水平降低。研究鉴定出TBL1x蛋白是NE在TAD中的一种新型作用底物和功能性下游靶标。功能缺失实验表明:NE通过调节TBL1x-LTA4H(白三烯A4水解酶)信号,介导炎症细胞的跨内皮迁移;同时,NE通过调控TBL1x-MECP2(甲基CpG结合蛋白2)信号轴,在TAD病理条件下调节平滑肌细胞的表型转换。进一步机制研究表明,抑制TBL1x会减少TBL1x和组蛋白去乙酰化酶3(HDAC3)分别与 MECP2 和 LTA4H 基因启动子的结合。最后,通过AAV2介导的主动脉平滑肌细胞 Tbl1x 基因敲低,证实了TBL1x在NE介导的TAD形成中的调控作用。
结论:本研究揭示了NE及其靶蛋白TBL1x在TAD中调控炎症细胞迁移和平滑肌细胞表型转换的关键作用。研究结果表明,NE-TBL1x信号轴是治疗高危TAD患者的一个有价值的作用靶点。
rId6
胸主动脉夹层(TAD)是一种致死性的主动脉病变,以主动脉内膜撕裂为特征,在65岁以上人群中的发病率约为35/100000,死亡率与致残率很高。尽管主动脉瘤(AA)和TAD具有多种共同的组织病理学改变,例如血管平滑肌细胞(SMC)因凋亡或坏死而消失、主动脉中层黏液样变性以及细胞外基质(ECM)破坏,但在大多数(>80%)TAD病例中并未观察到预先存在的动脉瘤。此外,动物研究表明,在载脂蛋白E(ApoE)缺陷小鼠中,TAD的发生先于血管紧张素II(Ang II)诱导的动脉瘤和动脉粥样硬化的形成,提示这两种致命性主动脉疾病存在不同的发病机制。虽然转化生长因子β(TGFβ)和Ang II信号失调、缺氧与氧化应激增加、基质金属蛋白酶(MMPs)与其内源性抑制剂(TIMPs)之间的失衡以及炎症反应均被认为参与了TAD的发病过程,但其确切的细胞与分子机制仍未完全阐明。
中性粒细胞弹性蛋白酶(NE)是一种丝氨酸蛋白酶,其主要已知功能是降解细胞内外的病原体。它对多种ECM蛋白以及非基质蛋白(如细胞因子/趋化因子、细胞表面蛋白/受体及其他功能性可溶性蛋白)具有较强的蛋白水解活性。NE与多种破坏性和炎症性疾病相关,包括慢性与急性肺部疾病以及心血管疾病。具体而言,在人类动脉粥样硬化斑块中可检测到NE的mRNA和蛋白,且动脉粥样硬化发展过程中血浆NE活性升高。我们及其他研究团队已证实NE在动脉粥样硬化发生及损伤诱导的新内膜SMC增生中具有致病作用。然而,关于NE是否参与TAD,目前所知甚少。本研究利用我们之前构建的NE与ApoE双基因敲除小鼠(ApoE⁻/⁻/NE⁻/⁻,简称NE-KO小鼠)及其同窝对照小鼠(ApoE⁻/⁻/NE⁺/⁺,简称WT小鼠,二者均为C57BL6/J背景),旨在确定NE在TAD发生与发展中的因果作用。同时,应用NE药理抑制剂GW311616A,探索抑制NE在治疗TAD方面的潜力。我们证明了NE在TAD形成与发展中起着关键作用,并发现含F-box样/WD重复序列的蛋白TBL1x(TBL1x)是NE在TAD环境下的功能性作用底物。
rId7
1 .NE 缺乏可防止 BAPN 诱导的 TAD 形成/发育
我们在之前的研究中生成了基因背景为 C57BL6/J 的 NE 缺失(NE-KO)小鼠及其对照同窝鼠(WT)。NE-KO 小鼠在整个生命周期中未观察到明显的整体表型,在生理条件下,NE-KO 小鼠显示出与 WT 对照同窝鼠相似的主动脉解剖特征(图 S1)。本研究利用广泛报道的通过在饮用水中添加 BAPN(0.25%,重量/体积)诱导的小鼠 TAD 模型来研究 NE 在 TAD 形成和发病中的潜在因果关系。最初,存活率、TAD 发生率(定义为死于胸主动脉破裂的小鼠和发现≥1 种主动脉病变[全厚主动脉内膜撕裂、穿透性主动脉溃疡、假腔和壁内血肿]的小鼠、 我们监测了雌雄 WT 小鼠的主动脉壁弹性纤维断裂情况和主动脉 NE 基因表达情况,以确定 BAPN 诱导的 TAD 形成的性别二态性程度(图 S2)。然而,我们并没有发现任何明显的表型差异(图 S2),这表明性别并不是 BAPN 诱导 TAD 形成的决定因素;因此,在接下来的研究中,我们将雌雄小鼠的所有数据集中起来进行统计分析。此外,一项时间进程研究显示,BAPN 诱导的 TAD 主要发生在服用 BAPN 后的第三周和第四周,在为期 4 周的研究方案结束时,累计 TAD 发生率为 85%(图 S2B)。在治疗的第二周偶尔也会观察到 TAD(<10%),在一些小鼠中还观察到硬膜内血肿(图 S3A 至 S3C)。重要的是,BAPN 治疗可显著提高主动脉 NE 基因表达水平(图 S3D)和血浆 NE 活性水平(图 S3E),并在 BAPN 给药后 2 周达到峰值。这些数据进一步表明 NE 参与了 TAD 的形成和发育。
为了证实这种作用,WT 和 NE-KO 小鼠都接受了 BAPN 给药。意料之中的是,NE-KO 小鼠的 NE 主动脉表达水平(图 1A)和血浆活性(图 1B)均不存在或极低,而 WT 小鼠服用 BAPN 则显著提高了它们的表达水平。重要的是,WT 小鼠和 NE-KO 小鼠在使用药物治疗时均未观察到死亡,但在为期 4 周的治疗方案中,使用 BAPN 治疗的 15 只 WT 小鼠中有 7 只死亡,14 只 NE-KO 小鼠中有 1 只因主动脉破裂而死亡(图 1C)。此外,与 WT 小鼠相比,用 BAPN 治疗的 NE-KO 小鼠的 TAD 发生率较低,弹性纤维断裂水平也有所下降(图 1D 至 1G),这证实了 NE 在 TAD 发生和主动脉破裂中的因果作用。
rId8
图 1. NE在BAPN诱导的TAD形成过程中起着关键作用。给三周大的载脂蛋白E-/-/NE+/+(WT)或载脂蛋白E-/-/NE-/-(NE-KO)雌雄小鼠随机注射载脂蛋白E-/-/NE+/+(WT)或载脂蛋白E-/-/NE-/-(NE-KO),在饮用水中加入 0.25% 的 BAPN(重量/体积),分别注射 2 周(A 和 B)或 4 周(C-G)。A、通过实时定量聚合酶链反应(RT-qPCR)分析胸主动脉基因表达。B、使用市售试剂盒(ab204730)测定血浆 NE 活性。C、Kaplan-Meier 存活率曲线。7/15 表示 15 只小鼠中有 7 只死亡。D、主动脉的宏观图像。收集胸主动脉并进行弹性蛋白范吉森(EVG)染色(E)分析。这里展示的是 TAD 发生率(F;n=10(0)表示 10 只小鼠中无一发生 TAD)和弹性蛋白断裂(G)的定量数据。TAD发生率由主动脉破裂死亡的小鼠和主动脉病变(主动脉内膜撕裂、假腔、壁内血肿)≥1个的小鼠定义。红色箭头表示 TAD 位置。此处显示的数据分别为 5 只(A 和 B 中为 5 只小鼠)或指定数量(C、F 和 G 中显示)小鼠的代表性数据(D 和 E)或平均值(±SEM)。A、B 和 G,*P<0.001(与载体相比),#P<0.001(与 WT 相比),2-way ANOVA,并进行 Tukey 分析的事后检验。C,*P<0.001(相对于车辆),#P<0.001(相对于 WT),对数秩(Mantel-Cox)检验。F,*P<0.001(相对于车辆),#P=0.001503(相对于 WT),χ2 检验
2 .药物抑制 NE 可防止 TAD 的形成和主动脉断裂
与我们之前的研究类似,我们给 C57BL6/J 基因背景的 WT 小鼠注射了 NE 药物抑制剂 GW311616A,以探索 NE 抑制在 BAPN 诱导的 TAD 形成中的治疗潜力。我们发现,虽然 GW311616A 能显著抑制血浆 NE 的活性,但 GW311616A 对其他几种蛋白酶的活性(包括 CG、胰蛋白酶和凝血酶)并无明显影响(图 2A)。在功能上,服用 GW311616A 能显著降低主动脉破裂导致的死亡率(图 2B),并降低 TAD 发生率和弹性蛋白破碎率(图 2C 至 2F)。这些数据共同证明,用 GW311616A 对 NE 进行药理抑制可抑制 TAD 的发生并防止主动脉破裂。
rId9
图 2:药物抑制 NE可减少BAPN诱导的 AD 形成。对在饮用水中摄入 BAPN 的三周大 ApoE-/-/NE+/+ (WT) 雌雄小鼠分别随机给予溶剂对照(溶剂)或 NE 抑制剂(GW311616A [GW])2(A)或 4(B-F)周。A、血浆 NE、CG(cathepsin G)、胰蛋白酶和凝血酶活性。B、动物存活曲线。7/18 表示 18 只小鼠中有 7 只死亡。C至F,HE染色(C)、弹性蛋白van Gieson(EVG)染色(D)的代表性图像,以及胸主动脉夹层(TAD)发生率(E,16/18表示18只小鼠中有16只发生了TAD)和弹性蛋白断裂(F)的定量数据。红色箭头表示 TAD 或内膜撕裂。此处提供的数据具有代表性(C 和 D),或分别为 6 只(A)或 11 只(载体)/14 只(GW)小鼠(F)的平均值(±SEM)(n=6 或 11/14 只小鼠)。*P<0.001(与载体相比,A 和 F 中的非配对 t 检验),P=0.0398(B 中的 log-rank [Mantel-Cox] 检验),*P<0.001(与载体相比,E 中的 χ2 检验)。
3 .NE 缺乏可减少 NETs 的形成
据报道,NE 在NETs的形成过程中起着至关重要的作用,而且我们之前发现 NE 介导的 NETs 参与了损伤诱导的动脉重塑。因此,我们有理由假设 NE 基因失活也可能在 BAPN 诱导的 TAD 形成过程中损害 NETs 的形成。事实上,瓜氨酸组蛋白 H3 和 MPO-2 代替 NET 的生物标志物的免疫荧光染色显示,与 WT 小鼠相比,NE-KO 小鼠剖开的主动脉中 NETs 的形成明显减少(图 S4A 和 S4B)。从 WT 或 NE-KO 小鼠体内分离的中性粒细胞在受到光稳定剂-1-肉豆蔻酸-13-乙酸酯刺激时也观察到类似现象(图 S4C 和 S4D)。综上所述,上述证据明确支持 NE 在 BAPN 诱导的 TAD 形成和发展过程中对 NETs 的形成所起的作用。
4 .NE 基因失活可减少主动脉炎症细胞的积聚,降低主动脉 MMP 的表达和活性
我们用不同的抗体对服用 BAPN 2 周的小鼠主动脉组织进行了免疫荧光染色分析。我们观察到,WT 小鼠主动脉中积累了大量 Ly6G+ 中性粒细胞和 Iba1+ 巨噬细胞,而 NE-KO 小鼠主动脉中观察到的炎症细胞要少得多(图 S5A 至 S5F)。此外,IF 染色数据显示 NE-KO 主动脉中 MMP-2 和 MMP-9 蛋白表达量显著下降(图 S5A 至 S5F)。重要的是,我们发现与 WT 小鼠相比,NE-KO 小鼠主动脉 MMP-2 和 MMP-9 的总含量和活性水平均明显降低(图 S5G)。同样,我们观察到,与 WT 小鼠相比,NE-KO 小鼠主动脉中炎症基因(MCP-1、IL-12β 和 IL-6)和粘附分子(ICAM1 和 VCAM1)的基因表达水平均显著降低(图 S5H)。
5 .TBL1x 蛋白是 NE 的新型底物
一项平行研究发现 NE 在 Ang II 诱导的腹主动脉 AA 中起作用(数据未显示),因此进行了腹主动脉蛋白质组学分析,以确定 NE 在此类主动脉病理学中可能的靶蛋白。如图 S6 所示,在输注 Ang II 2 周后,发现 NE-KO 主动脉中分别有 132 个和 24 个蛋白质显著上调和下调。火山图(图 3A)显示,在差异表达的蛋白质中,TBL1x(又称 TBL1)是 NE-KO 主动脉中对 Ang II 输注反应上调最明显的蛋白质。Western印迹分析也证实了输注BAPN 2周的NE-KO小鼠胸主动脉组织中TBL1x蛋白的上调(图3B和3C)。有趣的是,我们发现 WT 和 NE-KO 小鼠的胸主动脉 TBL1x 基因表达在 BAPN 给药后无明显变化(图 3D),这表明在 TAD 病理条件下 NE 对 TBL1x 的转录后调控。相反,NE-KO小鼠主动脉中的LTA4H(或LKHA4[白三烯A4水解酶])蛋白水平在BAPN处理下显著下调(图3A至3C)。与 TBL1x 不同,LTA4H 基因表达在 NE-KO 主动脉中也明显下降(图 3D)。重要的是,体外蛋白质消化试验数据显示,NE 可直接裂解 TBL1x,但不能裂解 LTA4H(图 3E)。这些数据共同表明 TBL1x 是 NE 在 TAD 中的新型底物之一。
rId10
图 3. 在NE-KO主动脉中观察到 TBL1x水平升高和 LTA4H表达水平降低。A,火山图。八周大的雄性WT)或 NE-KO 小鼠通过渗透微型泵(2004 型;Durect)注入生理盐水或 Ang II(每分钟 1000 纳克/千克)2 周。提取腹主动脉蛋白并进行无标记定量蛋白质组学分析。火山图显示 NE-KO 主动脉与 WT 主动脉中蛋白质表达水平的 P 值(-log10)与折叠变化(log2)的对比。B到D,分别从接受BAPN治疗2周的WT和NE-KO雄性小鼠胸主动脉中提取蛋白质和RNA,并分别进行Western印迹(B和C)和实时定量聚合酶链反应(RT-qPCR;D)分析。本文数据为 5 只小鼠(n=5)的代表值(B)或平均值(±SEM)(C 和 D)。*P<0.001(相对于 WT,非配对 t 检验);如果任何分析的 P<0.05,但 P>0.001, 精确的 P 值包含在相应的数字中。E、Tbl1x 和 LTA4H 体外消化。将重组人 TBL1x 和 LTA4H 蛋白(1600 ng)与活化的 HNE(人中性粒细胞弹性蛋白酶;100 ng)在检测缓冲液中孵育 30 分钟,然后分别用 TBL1x 和 LTA4H 抗体进行 Western 印迹分析。本文展示了 3 个独立实验(n=3)的代表性图像
6 .NE 通过调节 Tbl1x/Lta4h 信号轴,介导炎症细胞跨内皮迁移
我们之前已经证明,在 BAPN 诱导的 TAD 形成过程中,NE 基因失活可减少主动脉炎症细胞的聚集(图 S5A 至 S5F)。此外,用 SMA、Ly6G 和 NE 抗体对服用 BAPN 3 周的 WT 小鼠的主动脉组织进行 IF 染色分析表明,在主动脉介质层内和夹层部位有大量表达 NE 的 Ly6G+ 中性粒细胞(图 S7),推断 NE 在 BAPN 处理后炎症细胞迁移和招募到主动脉壁中的作用。事实上,跨内皮细胞迁移试验的数据显示,NE缺陷的中性粒细胞和单核细胞在MIP2或MCP-1刺激下的跨内皮细胞迁移能力分别下降(图4A),这证实了在TAD病理条件下,NE在炎症细胞迁移和招募到主动脉壁中发挥了重要作用。此外,IF 染色数据显示,TBL1x 在主动脉 CD68+ 细胞中高表达(图 S8)。重要的是,我们发现敲除 TBL1x 能显著增加 THP-1 单核细胞的跨内皮迁移,但当 NE 活性被 GW311616A 抑制时,这种促进作用消失了(图 4B),这表明 TBL1x 在 NE 介导的单核细胞迁移中起着功能性作用。
rId11
图 4. NE通过调节 TBL1x/LTA4H 信号轴介导炎症细胞跨内皮迁移。A、从WT或NE-KO小鼠体内分离骨髓中性粒细胞或单核细胞,并进行跨内皮细胞迁移试验。将小鼠内皮细胞(EC;C166 细胞)预培养到透孔插入物(孔径 8 微米)上形成 EC 单层,然后分别在 100 毫微克/毫升 MIP2(针对中性粒细胞)或 MCP-1(针对单核细胞)的作用下进行跨内皮细胞试验。B,THP-1 细胞对 MCP-1 的跨内皮迁移分析。用对照组(sh-NT [非靶向 shRNA])或 Tbl1x 基因特异性(sh-Tbl1x)shRNA慢病毒感染 THP-1 细胞,在没有或有 NE 抑制剂(GW311616A [GW],40 nM)的情况下进行跨内皮细胞迁移试验。C, 实时定量聚合酶链反应(RT-qPCR)分析显示 THP-1 细胞中 Tbl1x 和 Lta4h 基因的表达。D、TBL1x 通过调节 LTA4H 介导 THP1 细胞跨内皮迁移。用对照组(sh-NT)或基因特异性(sh-Tbl1x 或 sh-Lta4h)shRNA 慢病毒感染 THP-1 细胞,进行跨内皮细胞迁移试验。E至G,NE通过调节TBL1x/LTA4H信号轴介导单核细胞招募进入主动脉壁。用对照组(sh-NT)或 Tbl1x 基因特异性(sh-Tbl1x)shRNA 慢病毒感染从年龄和性别匹配的 WT 或 NE-KO 小鼠身上分离的骨髓单核细胞,并用BAPN诱导 2 周。对一部分单核细胞进行 RT-qPCR 分析(E),其余细胞用细胞追踪橙色标记(C2927;Invitrogen 公司),并通过尾静脉随机注射到预先注射 BAPN 2 周的年龄和性别匹配的 NE-KO 小鼠体内(每只小鼠 5×106 个细胞)。24 小时后,收集主动脉进行分析(F 和 G)。此处提供的数据为 5 个(n=5;A-D)或 6 个(n=6;E 和 G)独立实验/小鼠的代表值(F)或平均值(±SEM)。*P<0.001 (相对于 WT、溶剂或 shNT) 和 #P<0.001 (相对于 sh-NT);如果任何分析的 P<0.05 但 P>0.001, 精确的 P 值包含在相应的图中。A,非配对 t 检验。B 至 D、E 和 G,双向方差分析,并进行 Tukey 后检验。
有趣的是,由于 LTA4H 的主要功能是将白三烯 A4 转化为白三烯 B4,而白三烯 B4 是炎症性疾病中炎症细胞的一种强效趋化诱导剂,我们想知道 TBL1x 是否通过调节 LTA4H 来介导炎症细胞的迁移。为了解答这个问题,我们用 TBL1x 和 LTA4H 基因特异性 shRNA 慢病毒共同感染了 THP-1 单核细胞。不出所料,TBL1x 和 LTA4h 基因的表达受到了各自 shRNA 的特异性抑制(图 4C)。然而,我们观察到,虽然 TBL1x 基因敲除会显著上调 LTA4H 基因的表达,但并没有观察到 LTA4H 基因敲除对 TBL1x 基因表达的相互调控作用(图 4C),这证实了 LTA4H 是 TBL1x 的下游调控基因。功能研究表明,敲除TBL1x和LTA4H可分别显著提高和降低THP-1单核细胞对MCP-1刺激的跨内皮迁移能力,但敲除TBL1x对单核细胞迁移的促进作用被LTA4H抑制所减弱(图4D),这表明TBL1x/LTA4H信号在NE介导的炎症细胞跨内皮迁移中起着关键作用。
为了进一步证实 TBL1x 在 TAD 病理情况下 NE 介导的炎症细胞募集到主动脉壁中的体内作用,用对照组和 TBL1x 基因特异性 shRNA 慢病毒感染从 BAPN 处理的小鼠体内分离的 WT 和 NE 缺失型骨髓单核细胞。培养 24 小时后,通过尾静脉注射将骨髓单核细胞移植到用 BAPN 预处理 2 周的 NE-KO 小鼠体内。基因表达数据显示 NE-KO 骨髓单核细胞中 LTA4H 水平下降,而 Tbl1x 基因敲除可显著提高 WT 和 NE-KO 骨髓单核细胞中 LTA4H 的表达水平(图 4E)。因此,我们观察到,虽然 NE 基因缺失和 Tbl1x 基因沉默可分别显著减少和增加骨髓单核细胞向主动脉壁的募集,但当 Tbl1x 基因沉默时,NE 基因失活对骨髓单核细胞募集的抑制作用消失了(图 4F 和 4G)。值得注意的是,被回收转移的骨髓单核细胞在主动脉中的聚集可能受到多种因素的影响,如存活率和向其他组织(包括肺、肝脏和脾脏)的脱靶归巢。尽管如此,上述数据表明,在 TAD 病理学背景下,TBL1xLTA4H 信号轴至少是 NE 介导的主动脉壁炎症细胞招募的部分原因。
在 TAD 病理条件下,NE 通过调节 TBL1x/MECP2 信号轴,介导 SMC 表型调节
由于 SMC 表型转换或调节已被确定为 AA 和夹层形成和发展的基本机制之一,因此我们想知道 NE 介导的 AA 和夹层形成是否也有类似的机制。为了验证这一机制,我们首先检测了 NE 在主动脉细胞中的蛋白表达,发现 NE 在 TAD 病理条件下的主动脉 SMC 中高表达(图 S9A)。此外,我们还观察到 Ang II 增加了培养的主动脉 SMCs 中 NE 基因的表达,而加入 BAPN 后,NE 基因的表达呈剂量依赖性进一步显著上调(图 S9B)。我们还观察到 Ang II 和 BAPN 对主动脉 SMC 细胞裂解物(图 S9C)和条件培养基(图 S9D)中 NE 活性的类似调节作用。如前所述(图 S9A),IF 染色数据显示,在 BAPN 诱导的 TAD 形成过程中,TBL1x 蛋白也在主动脉 SMC 中高表达,在 NE-KO 小鼠中表达水平更高(图 5A;图 S10)。有趣的是,在 NE-KO 主动脉中观察到 SMC 特异性蛋白(SM22 和 calponin)表达水平较高(图 5B)。基因表达数据还显示,与 WT 对照组相比,NE-KO 主动脉中多个 SMC 特异基因的表达水平在 BAPN 处理后显著增加(图 5C),但在用车辆对照处理的 NE-KO 主动脉中未观察到这种差异(图 S11A),这推断 NE 在 TAD 病理条件下的 SMC 表型转换中起调控作用。
rId12
图 5. 在TAD病理条件下,NE通过调节 TBL1x/MECP2信号轴介导SMC表型调节。A、免疫荧光染色定量分析显示胸主动脉中 TBL1x 蛋白表达增加。B 和 C,给三周大的WT和NE-KO小鼠在饮用水(0.25% wt/vol)中添加BAPN2 周,提取胸主动脉蛋白和 RNA 并分别进行 Western 印迹(B)和RT-qPCR(C)分析。D和E,NE缺乏可防止Ang II/BAPN诱导的SMC表型转换。用 10 nM Ang II 和对照组或 25 µg/mL BAPN 处理血清饥饿的 WT 或 NE-KO SMC 48 小时。 收集总 RNA 和细胞裂解液并分别进行 RT-qPCR (D) 和 Western 印迹 (E) 分析。F,TBL1x 是 NE 介导的 SMC 基因调控所必需的。用对照组(sh-NT [非靶向 shRNA])或 Tbl1x(shTbl1x)型(shRNA)感染 WT 或 NE-KO SMC,并用 10 nM Ang II 和 25 µg/mL BAPN 处理 48 小时。收集总 RNA 并进行 RT-qPCR 分析。G和H,CHIP测定显示,抑制 TBL1x 会减少 TBL1x(G)和 HDAC3(H)分别与 MECP2 和 LTA4H基因启动子的直接结合。本文数据为 5 只小鼠(A)或 5 个独立实验(B-H,n=5)的代表值(B1 和 E1)或平均值(±SEM)。*P<0.001(相对于 WT、WT/Ctrl 或 WT/sh-NT)和 #P<0.001(相对于 WT/BAPN、NE-KO/sh-NT 或 GW311616A [GW]/sh-NT);对于任何分析的 P<0.05,但 P>0.001, 精确的 P 值包含在相应的数字中。A 至 C,非配对 t 检验。D 至 H,双向方差分析及 Tukey 后检验
由于我们以前曾报道 MMP-8 对 BAPN 诱导的 TAD 形成的促进作用是血管紧张素 II 生成增加的基础,我们想知道是否能在 NE 介导的 TAD 形成中观察到类似现象。虽然 BAPN 会显著增加血浆 Ang II 水平,但在 WT 和 NE-KO 小鼠之间未观察到差异(图 S11B)。此外,由于我们观察到 Ang II 和 BAPN 对 SMC 中 NE 的表达和活性有协同作用(图 S9B 至 S9D),我们研究了 Ang II 和 BAPN 对 SMC 基因表达是否有类似的协同作用。事实上,数据显示,单独使用 BAPN 对多种 SMC 特异性基因表达无明显影响(图 S11C),而在 10 nM Ang II 存在的情况下,BAPN 有显著的抑制作用(图 S11D),这表明 Ang II 和 BAPN 对 SMC 基因调控有协同作用。因此,在没有或有 BAPN 的情况下,WT 和 NE-KO SMC 与 Ang II 一起孵育,以进一步证实 NE 在 SMC 表型调节中的调控作用。我们观察到,与单独处理 Ang II 相比,Ang II/BAPN 联合处理可显著降低 WT SMC 的基因表达,但 NE-KO SMC 的基因表达却没有降低(图 5D),这在蛋白水平上得到了进一步证实(图 5E)。重要的是,TBL1x敲除可显著下调WT和NE-KO主动脉SMC的SMC基因表达(图5F),说明TBL1x在TAD病理条件下NE介导的SMC表型调控中起着关键的调控作用。
由于先前已发现 MECP2 是干细胞衍生的 SMC和主动脉 SMC中 SMC 基因表达的关键抑制因子,因此我们推测 MECP2 可能在 TAD 病理条件下 NE 介导的 SMC 表型转换中发挥作用。事实上,我们观察到 NE-KO 小鼠在接受 BAPN 治疗 2 周后,主动脉中 MECP2 基因(图 5C)和蛋白(图 5B)的表达水平均显著下降。此外,Ang II/BAPN 共处理可显著增加 WT 主动脉 SMC 的 MECP2 基因和蛋白表达,但 NE-KO 主动脉 SMC 的 MECP2 基因和蛋白表达却没有增加(图 5D 和 5E)。有趣的是,在 WT 和 NE-KO SMC 中,敲除 TBL1x 会显著上调 MECP2 基因的表达(图 5F),这表明 MECP2 受 TBL1x 的负调控。重要的是,BioGRID(相互作用数据集生物总库)网络(https://thebiogrid.org/112770)预测 TBL1x、MECP2 和 HDAC3 之间存在密切的相互作用(图 S12),这表明 HDAC3 在 TBL1x 介导的基因调控中起着调控作用。事实上,染色质免疫沉淀试验的数据显示,虽然在 NE-KO SMCs 中观察到 TBL1x 和 HDAC3 与 MECP2 基因启动子的结合增加,但敲除 TBL1x 会显著抑制这种结合(图 5G 和 5H,右图)。在 THP-1 单核细胞中,TBL1x 和 HDAC3 与 LTA4H 基因启动子的结合也有类似的效果(图 5G 和 5H,左)。这些数据共同证实了 TBL1x/HDAC3/ MECP2 信号在 TAD 病理条件下 NE 介导的 SMC 表型调节中的调控作用。
8 .抑制 Tbl1x 可削弱 NE 基因缺陷-对 BAPN 诱导的主动脉夹层形成的保护作用
为进一步探索 TBL1x 在 NE 介导的 SMC 表型调节和 TAD 中的潜在作用,我们在 WT 和 NE-KO 小鼠体内进行了一项研究,使用 AAV2 载体在主动脉 SMC 中进行特异性基因转移和敲除,如前所述,如图 S13 和 S14 所示。基因表达谱数据显示,虽然 NE-KO 小鼠主动脉中 MECP2 基因表达显著下降,但抑制 Tbl1x 基因可显著上调 WT 和 NE-KO 小鼠的 MECP2 基因表达。重要的是,在所有 SMC 基因中都观察到了相反的趋势(图 6A),证实了 TBL1x 在 TAD 病变中对 MECP2 和 SMC 基因表达的调控作用。从表型上看,注射了 AAV2-sh-Tbl1x 的 WT 小鼠死亡率更高,而 WT/ AAV2-sh-NT 和 NE-KO/AAV2-sh-Tbl1x 小鼠的死亡率水平相当(图 6B)。此外,我们还观察到,注射 AAV2-sh-NT 后,NE-KO 小鼠的 TAD 发生率较低,弹性纤维断裂程度也有所下降,但在 WT 和 NE-KO 小鼠中,Tbl1x 基因沉默后,TAD 发生率和弹性纤维断裂程度都较高(图 6C 至 6E;图 S15)。这些数据共同表明,TBL1x/MECP2 信号的调节是 NE 介导的 TAD 形成的主要潜在机制之一。
rId13
图 6. 抑制 Tbl1x可减弱 NE基因缺陷对BAPN诱导的 AD 形成的保护作用。如图 S13A 所示,给三周大性别匹配的WT或NE-KO小鼠随机注射非靶向对照(sh-NT [非靶向 shRNA])或 Tbl1x 基因特异性(sh-Tbl1x)shRNA腺相关病毒-2(AAV2),然后注射 BAPN。分别在 1 周(A)或 4 周(C-E)收集胸主动脉,然后进行各种分析。A、通过RT-qPCR分析检测的主动脉基因表达水平。B、动物存活曲线(每组 15 只小鼠)。C至E,弹性蛋白范吉森(EVG)染色的代表性图像(C),以及TAD发生率(D,12/15表示15只小鼠中有12只发生了TAD)和弹性蛋白断裂(E)的定量数据。红色箭头表示 TAD 或内膜撕裂。此处提供的数据分别为 5 只(A;n=5)或 6 至 13 只(C-E,n=6-13 只)小鼠的代表性数据(C)或平均值(±SEM)。*P<0.001(相对于 WT)和 #P<0.001(相对于 sh-NT);如果任何分析的 P<0.05,但 P>0.001, 精确的 P 值包含在相应的数字中。A 和 E,双向方差分析与 Tukey 分析的事后检验。B,对数秩(Mantel-Cox)检验。D,χ2 检验。
9 .观察到急性 TAD 患者体内 NE 增加
到目前为止,我们已经证明了 NE 在小鼠 AA 和夹层发病及主动脉破裂中的关键作用。H&E和弹性蛋白 van Gieson 染色的数据证实了 TAD 的病理特征,包括弹性层紊乱,弹性蛋白断裂频繁,弹性纤维耗竭,以及夹层人动脉中 SMC 的缺失,而正常健康的主动脉中未观察到此类病理特征(图 S16)。用 SMA 和 NE 抗体进行 IF 染色显示,NE 在断裂的人体主动脉内的 SMC 中高度表达(图 7A),与正常健康的人体主动脉相比,断裂的主动脉中 NE 蛋白表达量明显更高(图 7A 和 7B)。同样,在急性 TAD 患者中也观察到主动脉 NE 基因表达增加(图 7C)。此外,我们还观察到急性 TAD 患者的血清 NE 水平升高(图 7D),这证明 NE 可能参与了人类 TAD 的发病机制。重要的是,接收者操作特征曲线显示血清 NE 水平对人类急性 TAD 具有良好的诊断能力,接收者操作特征曲线下面积为 0.7415(图 7E)。具体而言,将血清 NE 水平的临界值设定为 2.783 纳克/毫升时,我们观察到急性 TAD 患者与正常健康人的鉴别特异性为 81%,灵敏度为 72%(图 7E)。
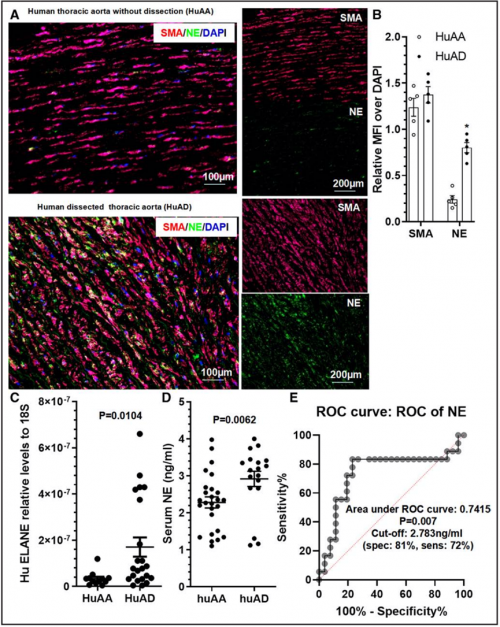
图 7. 有(HuAD)或无(HuAA)急性TAD的人胸主动脉中NE的检测。A和B,免疫荧光(IF)染色显示夹层人胸主动脉中NE蛋白表达水平升高。这里展示了 NE 或 SMA相对于 DAPI染色的代表性图像(A)和相对平均荧光强度(MFI;B)。*P<0.001 (n=5, vs HuAA; unaired t test)。C、RT-qPCR分析显示,夹层的人胸主动脉中 NE 基因表达增加。P=0.0104(HuAA 的 n=12 或 HuAD vs HuAA 的 n=22; Mann-Whitney U 检验)。D,ELISA 分析显示急性 TAD 患者血清 NE 水平升高。P=0.0062(与 HuAA 相比,HuAA 为 26 人,HuAD 为 18 人;Mann-Whitney U 检验)。E、显示血清 NE 水平对人类急性 TAD 诊断能力的接收操作特征曲线(ROC)。
rId15
TAD 是一种危及生命的急症,因主动脉破裂而导致的死亡率和发病率都很高。由于对 TAD 的病因缺乏新的认识,目前还没有有效的药物治疗方法来延缓 TAD 的发生和预防主动脉破裂。在这里,我们证明了 NE 在促进 TAD 的形成和发展中起着至关重要的作用。具体而言,通过基因缺失和药物抑制 NE 可显著降低 TAD 的发病率,并通过防止主动脉破裂降低死亡率,从而使 NE 成为治疗 TAD 发病和主动脉破裂高危患者的一种有价值的疗法。
在本研究中,我们将 0.25%的 BAPN加入 3 周龄小鼠的饮用水中,观察到 TAD 发病率和死亡率分别下降了 85% 和 46.7%。我们的研究结果与之前多项研究中的观察结果一致,但与另一项研究中的一些观察结果略有不同。这种差异可能是由于这些研究中用于评估 TAD 发生率的实验环境和参数不同造成的。例如,我们的研究中使用的是 3 周大的小鼠,而他们的研究中使用的是成年小鼠(10-15 周大)。此外,虽然这两项研究中使用的小鼠都具有相同的 C57BL/6J 遗传背景,但我们的研究中使用的小鼠还具有载脂蛋白 E 基因缺失,而载脂蛋白 E 基因缺失是众所周知的多种主动脉病变的影响因素。重要的是,Qi 等人使用 BAPN 和 Ang II 联合给药诱导 AA/主动脉夹层,而我们仅给小鼠注射 BAPN 来诱发 TAD。最后,在我们的研究中,TAD发生率的定义是因胸主动脉破裂而死亡的小鼠,以及发现≥1种主动脉病变(主动脉内膜撕裂、穿透性主动脉溃疡、假腔和壁内血肿,壁内血肿是指在弹性层间发现红细胞或血栓)的小鼠,同时小鼠显示出其他主要主动脉病变,包括层间间隙增宽、 值得一提的是,我们可能会遗漏一些因膜内血肿消退而导致主动脉夹层不明显的 TAD 病例,这也是我们研究的局限性之一。
我们以前曾报道过 NE 在动脉粥样硬化和损伤诱导的新内膜形成中的关键作用。具体而言,我们发现 NE 可通过增加全身和主动脉炎症来促进动脉粥样硬化并增加斑块的易损性。NE 通过增加 ATP 结合盒转运体 ABCA1 蛋白降解和抑制巨噬细胞胆固醇外流,从而促进泡沫细胞的形成,发挥其促炎症作用。此外,最近通过多种方法证实了 NE 在损伤诱导的新生内膜形成中的功能性作用,以及以 NE 为靶点治疗血管成形术后再狭窄的治疗潜力。我们的研究表明,NE 可促进 SMC 增殖、迁移和炎症反应,NE 通过调节 TLR4 / MyD88 / IRAK1 / TRAF6 / NF-κB 调控轴介导这些 SMC 功能障碍表型和损伤诱导的新内膜 SMC 增生。有趣的是,Eliason 等人首次报道了循环中性粒细胞及其对主动脉壁的浸润是主动脉NE灌注诱导实验性 AA 形成的重要初始触发因素,中性粒细胞耗竭可抑制 AA 的发展,这表明中性粒细胞在 AA 形成中发挥着重要作用。此外,DPPI–一种溶酶体半胱氨酸蛋白酶,对激活中性粒细胞颗粒储存的丝氨酸蛋白酶(包括 NE)至关重要–被报道NE诱导的实验性 AA 的形成过程中起着至关重要的作用,它能促进中性粒细胞被募集到NE酶损伤的主动脉壁,并增加 CXC 趋化因子配体 2 的局部生成。重要的是,同一研究小组后来的一项研究表明,缺少 2 种中性粒细胞丝氨酸蛋白酶(NE 和蛋白酶-3)可再现 DPPI 缺失小鼠的 AA 抗性表型,从而推断 NE 在 AA 的发生发展中起着重要作用。此外,药物抑制弹性蛋白酶的活性可防止弹性蛋白酶输注诱导的实验性腹部 AA 的发展。关于炎症细胞可能参与 TAD 的问题,有报道称,有针对性地消耗小鼠体内的单核细胞/巨噬细胞可抑制 TAD 的发生,这证实了这些炎症细胞在 TAD 的发展中起着关键作用。此外,在因急性 TAD 而接受急诊手术的患者中,手术期间静脉注射 NE 抑制剂西维来司他钠可恢复术后抗凝血酶 III 和血小板水平。这一干预措施还缩短了术后机械通气的时间,表明抑制 NE 对接受急诊手术的急性 TAD 患者有一定的益处。然而,上述研究主要使用NE输注诱导的实验性 AA 来研究中性粒细胞丝氨酸蛋白酶在这种主动脉疾病中的潜在参与,这在一定程度上阻碍了我们对内源性 NE 对 AA 形成和主动脉夹层发生/发展的因果关系的了解。在本研究中,我们首先发现 NE 的表达和活性在 BAPN 诱导的 TAD 和急性 TAD 患者中均有所增加。重要的是,基因缺失和药物抑制实验的数据证实了内源性 NE 在 TAD 发病和进展以及主动脉破裂中的因果作用,并令人信服地证明了 NE 抑制剂在治疗 TAD 患者方面的治疗潜力。
有趣的是,我们的数据显示,在 BAPN 诱导的 TAD(图 S7、S8、S9A 和 S10)和解剖的人体动脉(图 7A)中,除中性粒细胞外,NE 在巨噬细胞和 SMC 等其他细胞中也有高表达,这与我们之前在动脉粥样硬化斑块和损伤动脉等其他病理情况下的观察结果一致。在 Ang II 和 BAPN 的联合作用下,主动脉 SMCs 中 NE 的浓度显著升高,细胞裂解物和细胞培养上清液中检测到较高的 NE 酶活性证明 SMCs 能产生和分泌 NE(图 S9B 至 S9D)。这些结果表明,在 TAD 的体内疾病环境中,多种细胞(如中性粒细胞、巨噬细胞和 SMC)可通过自分泌或旁分泌的方式相互作用并调节彼此的功能。事实上,我们观察到大量表达 NE 的中性粒细胞被招募/迁移到介质层和解剖部位(图 S7)。因此,我们推测这些被招募的炎症细胞以及被激活的常住 SMCs 产生/分泌了 NE,而 NE 可直接裂解/降解 ECM 蛋白(如胶原、弹性蛋白、糖蛋白),从而削弱主动脉壁。另一方面,产生/分泌的 NE 也可能作用于 SMC,导致 SMC 调节失调,从而促进 TAD 的发生。然而,值得一提的是,本研究的局限性之一在于我们目前的数据无法全面阐述不同细胞 NE 来源形成 TAD 的可能性。此外,NE 作为一种分泌蛋白的特性也使这种划分变得极为困难,因为这种蛋白可以通过自分泌和旁分泌两种方式发挥其细胞功能。例如,我们以前曾报道过,血管内膜干细胞/祖细胞不表达 MMP-8,而是吸收巨噬细胞分泌的 MMP-8,进而促进其向 SMCs 分化,从而促成损伤诱导的新内膜形成。
从机理上讲,我们已经提供了明确的证据支持以下观点,即在 TAD 病理条件下调节 NETs 的形成(NETosis)是 NE 介导的 TAD 形成/进展的分子机制之一。研究发现,NETosis 发生在实验性 AA 的早期阶段(早在动脉瘤诱导后 2-3 天),并且已被广泛报道为 AA 形成和发展的关键驱动因素和决定因素之一。重要的是,利用 DNase I 降解和抑制 NETosis,成功证实了在NE诱导的实验性 AA 中靶向 NETosis 的治疗潜力。与这些观察结果一致的是,我们在本研究中观察到,在 TAD 病理条件下,WT 而非 NE-KO 主动脉和中性粒细胞中存在丰富的 NETosis,这可能是 NE 缺失小鼠和接受 NE 抑制治疗的 WT 小鼠中观察到的 TAD 抗性表型的原因。
具体而言,研究人员利用复合缺陷小鼠和巨噬细胞输注,证明巨噬细胞源性 MMP-9 和间质细胞 MMP-2 都是产生 AA 的必要条件和协同作用。有趣的是,多项研究表明,在各种实验条件下,NE 在调节和激活 MMP-267 和 MMP-968 方面起着调节作用。在肿瘤侵袭和血管生成、急性肺损伤或单核细胞和成纤维细胞在三维胶原凝胶中的长期培养过程中,NE 被认为是 MMP-2 和 MMP-9 的生理性激活剂。此外,有报道称,在实验性腹膜炎模型中,NE 活性的增加可弥补 MMP-9 基因失活对白细胞浸润的调节作用。此外,NE 还与人类囊性纤维化蛋白溶解功能障碍有关,这体现在 NE 能单独裂解和激活 MMP-9,但却能降解组织金属蛋白酶抑制剂-1。上述研究结果与我们的观察结果非常吻合,即在 BAPN 处理下,NE-KO 主动脉中 MMP-2 和 MMP-9 的表达水平和活性均显著下降(图 S5)。因此,我们可以得出这样的结论:主动脉中 MMP-2/9 的表达或活性随着 BAPN 处理的增加而增加是 NE 介导的 TAD 发生和发展的另一种潜在分子机制。
此外,本研究的一个重要而新颖的发现是,我们已确定 TBL1x 是 NE 在 TAD 中的功能性底物和靶点,也是 NE 促进 TAD 形成和发展的一个关键下游信号分子。具体来说,我们提供了几项证据来支持这一结论。首先,在我们的主动脉蛋白质组学分析中,TBL1x 被确定为 NE-KO 主动脉中对 Ang II 输注反应调节最显著的蛋白质。其次,我们发现 NE-KO 小鼠在接受 BAPN 治疗后,主动脉 TBL1x 蛋白表达水平明显增加,但其 mRNA 表达水平却没有增加。第三,我们的体外蛋白质消化数据提供了直接证据,表明 TBL1x 蛋白可被 NE 直接裂解和降解。进一步的多种生化和生物学实验证实,在炎症细胞跨内皮细胞迁移和血管SMC表型调控方面,TBL1x是NE的一个功能性下游靶点。最后,重要的是,在 BAPN 处理下,AAV2 介导的 Tbl1x 基因抑制主动脉 SMC 后,在 NE 缺失小鼠中观察到的 TAD 抗性表型消失了,这表明 TBL1x 在 NE 介导的 TAD 发生和发展中起着关键作用。
后来的一项研究表明,TBL1x 是配体核受体和 PPARγ诱导的胚胎干细胞成脂分化所介导的转录激活所必需的。此外,研究还发现 TBL1x 在 Wnt-β-catenin 信号转导中发挥调控作用,其方式是直接与 β-catenin 相互作用并将其招募到细胞核或保护 β-catenin 免受 Siah-1泛素化。重要的是,研究表明 MECP2 可直接与 TBL1x 及其相关蛋白 TBLR1 结合,而这种相互作用对优化脑功能至关重要。有趣的是,我们先前已证实 MECP2 是 SMC 分化和表型调节的关键抑制因子。事实上,我们发现在 Ang II/BAPN 诱导的 SMC 表型转换过程中,MECP2 基因的表达受到 TBL1x 的负调控,而且我们的研究结果证实 TBL1x 可直接与 MECP2 基因启动子结合。因此,我们在本研究中提供了明确的证据,支持 TBL1x 通过抑制 SMC 中 MECP2 的表达来维持 TAD 病理条件下 SMC 收缩表型的观点。
值得注意的是,NE 抑制和基因失活对 TAD 形成和发病的潜在影响可能并不完全归因于 TBL1x 和基质纤维的降解,NE 介导的 TAD 发生的潜在分子机制可能并不局限于本研究中考察的部分。如图 3A 和图 S6 所示,研究发现主动脉中还有 154 种蛋白质受 NE 基因失活的影响。因此,这些被调控的蛋白可能代表了 NE 促进 TAD 发生和进展的其他可能靶点。其中,IRGM1、CD180、FBN1和 THSD4尤其引起注意。具体而言,多项遗传学研究发现了 THSD4 和 FBN1 基因中的多种致病变异,并显示这些致病变异与 AA/TAD 有因果关系,但与其他血管疾病无关,这就指出了这两种蛋白在 AA/TAD 中的功能性作用。此外,CD180已被确认可与 TLR4 合作介导先天性免疫反应和 NF-κB 激活。有趣的是,我们先前已确认 TLR4 是 NE 介导主动脉炎症的功能靶点,从而推断 CD180/TLR4 信号传导在 NE 介导的主动脉炎症和 TAD 发展中的潜在作用。此外,IRGM1 被广泛报道可调控炎症细胞的运动、巨噬细胞的极化和自身免疫,所有这些都可能导致 TAD 的形成。因此,这些蛋白在 NE 介导的 TAD 发生和进展中的功能影响值得进一步研究。
总之,我们揭示了 NE 在 TAD 形成/进展中的关键作用,并证明 NE 可通过加强炎症细胞向血管壁的招募/迁移、增加主动脉炎症和促进 SMC 表型转换来促进 TAD 的发生和主动脉破裂。NE 至少部分通过直接裂解和降解 TBL1x 蛋白来发挥上述细胞功能,而 TBL1x 蛋白可抑制 LTA4H 信号传导,进而增强炎症细胞跨内皮细胞迁移,从而加剧主动脉炎症。同时,TBL1x 的减少也会抑制 MECP2 的表达,进而促进 SMC 向去分化表型转变。TBL1x 通过增加 HDAC3 与 MECP2/LTA4H 基因启动子的直接结合来抑制 MECP2 和 LTA4H 的表达。正如之前所报道的,NE 还可通过调节 TLR4/NF-κB 信号通路增加主动脉和 SMC 的炎症反应。重要的是,NE 还可直接上调并激活 MMP-2 和 MMP-9,促进 NETosis。因此,NE 可通过多种信号通路(包括但不限于 MMP-2/9 激活、主动脉炎症、SMC 合成表型转换和 NETosis)促进 TAD 的形成。重要的是,本研究还强调了抑制 NE 对 TAD 患者的治疗潜力。
参考文献:
Yang M, Zhou X, Pearce SWA, Yang Z, Chen Q, Niu K, Liu C, Luo J, Li D, Shao Y, Zhang C, Chen D, Wu Q, Cutillas PR, Zhao L, Xiao Q, Zhang L. Causal Role for Neutrophil Elastase in Thoracic Aortic Dissection in Mice. Arterioscler Thromb Vasc Biol. 2023 Oct;43(10):1900-1920. doi: 10.1161/ATVBAHA.123.319281. Epub 2023 Aug 17. PMID: 37589142; PMCID: PMC10521802.
本文转载自独立学术期刊,仅供专业人士学术参考,不可作为诊疗依据,任何用药请严格遵循药品说明书.